
What are Bioequivalence Studies?

What are they?
What are bioequivalence studies? (BE) studies are a special type of studies designed to compare the (i.e the amount of drug that enters systemic circulation) of two products that are using the same active ingredient or molecule and to verify how similar they are to each other.
Once a pharmaceutical company develops a new drug (i.e. an innovator drug) which is based on a newly discovered active ingredient or molecule, that company will apply for a patent that will give it the complete control and production of that molecule. However, the patent only lasts for a certain amount of time (20 years in Canada for example). , the pharma company must demonstrate the drug’s safety and efficacy through clinical studies of Phase I, II and III. The aim of the company is to get their drug on the market as quickly as possible so that the marketed drug can be under patent protection as long as possible. That way, the pharma company has no competition for that specific drug and can generate revenues quicker.
Once that patent expires, other pharmaceutical companies can start producing their own version of the innovator drug (i.e. a generic drug). To be marketed, a generic drug must have the same effects (i.e. same absorption, distribution, metabolism and excretion) as the innovator one. In other words, they must be bioequivalent.
How are Bioequivalence studies performed?
In its simplest form, a bioequivalence study is performed as a 2×2 crossover as presented in the table below.

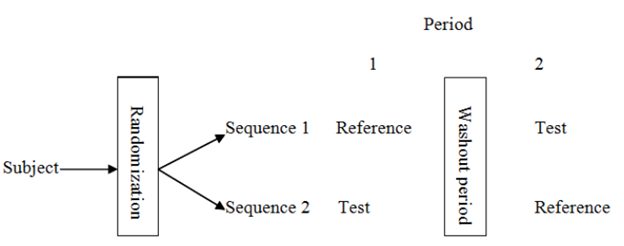
In this form, each enrolled subject is being administered the two drugs over different periods. Each subject is randomized to either one of two sequences that will determine which drug is taken first. This is to make sure that the drug intake order does not have an impact on the subject’s response. If all subjects took the Test before the Reference, the response could be different than if all subjects took the Reference before the Test. By randomizing each subject to a certain sequence, we are trying to control and reduce such effect (or bias).. A washout period is also included in the design. This is a period of time between the two periods in which the subject does not come to the clinical site. The duration of this period is usually 1-2 weeks, depending on the PK property of the drug. This period serves as a way to make sure that any amount of the treatment taken in Period 1 is eliminated and will not interfere with the response to the treatment taken in Period 2.
Since every subject is unique, each of them will have a different response to the two drugs based on a variety of factors. In a parallel design, each subject takes only one of the two drugs. This means that if subject have a higher response to a drug due to genetic reasons. It is not easy to know whether it is because of the drug or the subject itself. However, in a cross-over study design, the big advantage is that each subject acts as its own control. In other words, unlike a parallel design where, the effect of a drug on each subject is reduced to the fact that they all are taking the two drugs. We therefore have less subject related bias when we want to compare the response to the two formulations.
Note that the following example can be expanded to a 3×3, 4×4 etc. design if there are more different test or reference formulations to be tested. For a certain n x n design, each subject will take the n different formulations over n periods with a washout period between each treatment administration.
What are we looking for?
Compare how similar the two formulations are, we are looking at the amount of active ingredient in the blood or plasma. For example, the active ingredient in common pain medications is ibuprofen. To measure the amount, we use the concentration of active ingredient in the blood or plasma. The figure below, shows an example of how much active ingredient is in the blood after administration.
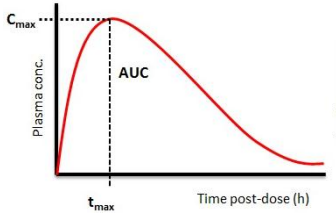
We can see that, at first, the concentration of active ingredient in the plasma goes up quite quickly until it reaches a maximum concentration or Cmax. Afterward, the concentration slowly goes down until it is almost not present anymore. The time at which the Cmax is attained is called Tmax. This type of curve will be observed for each subject and each formulation. We want those curves to be similar between the generic and innovator treatment. If we have that and after some statistical calculations, then we can say that the two treatments are similar or bioequivalent.
How many subjects are required?
Depending on various factors, the minimum sample size for each study varies. However, for any study of this type, the FDA requires that a minimum of 12 subjects are required for any BE studies (that is, at least 6 subjects per sequence if it is a 2×2 crossover design).
What type of subjects are we enrolling?
Usually, healthy subjects are preferred for BE studies to avoid any adverse events caused by the drug to a certain medical condition. It is also preferred to have male and female subjects from various ages and race to represent the whole population in which the drug will be marketed.


